
AMPAC Fine Chemicals (AFC), an SK pharmteco company, is a United States-based, custom manufacturer of active pharmaceutical ingredients (APIs, Drug Substances) and registered intermediates. AFC solves problems through technology and innovation to reliably deliver quality products that save and improve lives. With 80 years of experience, AFC has mastered challenging chemistries, enabling us to provide the highest quality services to our customers.
Our fully CGMP-compliant facilities located in California, Texas, and Virginia specialize in process development, scale-up, and production from kilograms to multi-ton quantities. AFC has expanded its capabilities to include contract analytical services. These services are conducted at AMPAC Analytical, located near our headquarters in California.
Blog
In the entries below, we’ll share our thoughts and comments on these areas and other topics of interest. Please feel free to contact us to suggest topics of interest.
8/29/2023
Nitrosamines: An Update
 The linkage between nitrosamines and cancer was first postulated by William Lijinsky in 1970. Then, in 2018, N-nitroso-dimethylamine (NDMA)) was detected in an active pharmaceutical ingredient, Valsartan (an Angiotensin-II-receptor antagonist). Finally, the FDA issued a guidance for the industry, “Control of Nitrosamine Impurities in Human Drugs”, in the fall of 2020. However, the guidelines continue… (Read More)
The linkage between nitrosamines and cancer was first postulated by William Lijinsky in 1970. Then, in 2018, N-nitroso-dimethylamine (NDMA)) was detected in an active pharmaceutical ingredient, Valsartan (an Angiotensin-II-receptor antagonist). Finally, the FDA issued a guidance for the industry, “Control of Nitrosamine Impurities in Human Drugs”, in the fall of 2020. However, the guidelines continue… (Read More)
8/11/2023
Dynamic Vapor Sorption
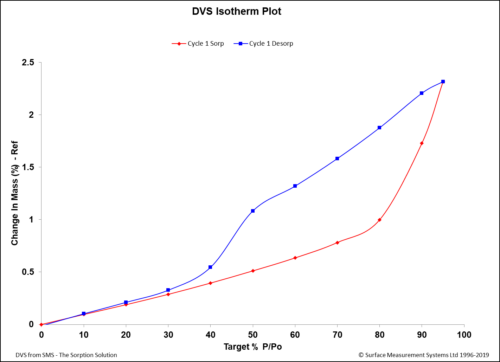
Dynamic Vapor Sorption (DVS) is a gravimetric technique used to measure the change in mass of a material in response to changes to surrounding conditions such as temperature or humidity. DVS is primarily used with water vapor but can be applied to other organic solvents as well for the physicochemical characterization of solids.
DVS was developed by… (Read More)
7/25/2023
Keys to Effective Method Development
 Effective method development is crucial for the quality control of Active Pharmaceutical Ingredients (API) and Drug Products (DP). Thorough method development enables successful downstream method validation.
Effective method development is crucial for the quality control of Active Pharmaceutical Ingredients (API) and Drug Products (DP). Thorough method development enables successful downstream method validation.
The regulatory guidance specifies that:
- The scope of development varies by application (quantitative, qualitative, etc.).
- It is phase appropriate.
- The client may provide additional guidance/validation criteria.
- The validation guidance directs how AMD (analytical method development) is conducted.
Early Adoption of Forced Degradation Analysis
It is recommended that forced degradation be performed early in the method development lifecycle and that method parameters are suitable for mass spectrometry. This will prevent… (Read More)
7/10/2023
It’s Always Time to Talk About Phase-Appropriate Method Development

The Cost of Drug Discovery and Development and How to Mitigate It
The Cost of Drug Discovery and Development and How to Mitigate It
The path to successful drug discovery and development is extremely long, expensive, and risky. It can take between 10 to 15 years at an average cost of more than $1–2 billion for every new drug approved for clinical use.1,2 In fact, preclinical drug discovery alone “typically takes five and a half years and accounts for about one-third of the cost of drug development.”3,4 Therefore, even during the earliest stages of a drug product or active pharmaceutical ingredient project, phase-appropriate method development should be instituted... (Read More)
6/20/2023
TCB* With Your TTC Needs

“The dose makes the poison” – Paracelsus (c. 1493– 1541), born Theophrastus von Hohenheim
The Threshold of Toxicological Concern (TTC) refers to levels of mutagenic impurities expected to pose a negligible carcinogenic risk.1 The US FDA, the EMA (European Medicines Agency), and the European Food Safety Authority (EFSA) all have TTC values and regulations in place for food and active pharmaceutical ingredients (APIs), along with numerous other products.2,3 Originally, these standards were applied to TTC levels from oral ingestion but have expanded to even include cosmetics and fragrances.4,5... (Read More)
6/7/2023
Forced Degradation Studies Can Reduce Stress(ors)
 Forced Degradation is an important addendum to our previous post on Stability and Storage. Stressors are applied to new APIs and drug products to determine their degradation pathways and products under a variety of environmental conditions, including acid, base, light, heat, and oxidation. Forced degradation studies are also known as stress testing, stress studies, stress decomposition studies, and forced decomposition studies. These conditions “…are more severe than accelerated (stability) conditions and thus generate degradation products that can be studied to determine the stability of the molecule.”1 ... (Read More)
Forced Degradation is an important addendum to our previous post on Stability and Storage. Stressors are applied to new APIs and drug products to determine their degradation pathways and products under a variety of environmental conditions, including acid, base, light, heat, and oxidation. Forced degradation studies are also known as stress testing, stress studies, stress decomposition studies, and forced decomposition studies. These conditions “…are more severe than accelerated (stability) conditions and thus generate degradation products that can be studied to determine the stability of the molecule.”1 ... (Read More)
5/8/2023
The Background, Advantages of, and Considerations for Radiolabeled Peptides
The use of radiolabeled peptides is a well-established tool in researching and treating many diseases and conditions. Selective receptor-targeting peptides are utilized as agents due to their rapid circulatory and tissue clearance and the high affinity and specificity to their targets. Peptides also have a relatively small size and low molecular weight compared to proteins and antibodies. There have been innovations and improvements in the design of peptides that incorporate chemical modifications with “impressive diagnostic accuracy and sensitivity.”1 Coupling these peptides with radiolabeling for peptide receptor radionuclide imaging (PRRI) and therapy (PRRT) has yielded remarkable results. In fact, a historical summary of radiolabeled peptides asserts, “The emergence of radiolabeled peptides for use with PET/CT such as 68Ga, 18F, and 64Cu, and the use of new receptor binding ligands…, have revolutionized PRRI and improved its diagnostic power beyond expectation.”2... (Read More)
4/25/2023
Raw Materials Testing: Trust – and Verify – Your Sources
The CGMP guidance for APIs from the FDA states that raw material specifications should be established and documented. The guide’s key line states, “Quality measures should include a system for testing raw materials, packaging materials, intermediates, and APIs. (19.23)”1

All raw materials used in producing APIs for clinical trials must be evaluated by testing or received from the supplier with accompanying analysis and subsequently subjected to identity testing. Raw materials and intermediates need to be designated by names and/or specific codes so that any special quality characteristics can be readily identified. Furthermore, written procedures should provide for the identification, documentation, appropriate review, and approval of any changes to raw materials. Additionally, changes to supply sources of critical raw materials should be treated according to the FDA’s established change control guidelines.…(Read More)
4/4/2023
Extractables and Leachables
Extractables and Leachables (E&L) are essential areas of concern for the pharmaceutical and food industries, specifically regarding their packaging, usage components (e.g., medical devices or syringes), and the manufacturing chain. We will examine testing of analysis of them within pharmaceutical applications. The two terms are related but distinct, each with its own analytical requirements.

A handy article published in Pharmaceutical Engineering by the International Society for Pharmaceutical Engineering (ISPE) explains that “Extractables are chemical compounds that migrate from single-use systems (SUS) into model solvent solutions under controlled and exaggerated conditions depending on temperature, pH, polarity, and time.” In other words, this happens when using strong solvents. They note that “SUS are normally not exposed to such conditions in biopharmaceutical processes.”1
ISPE’s article defines leachables as “chemical compounds that migrate from SUS into process solutions under normal biopharmaceutical process conditions. They further clarify that these compounds “may end up in the final drug product formulation. For the most part, leachables are a subset of extractables, although interaction with product components may produce leachables not seen as extractables.”1 …(Read More)
3/14/2023
Elemental Impurities
A special thanks to Matt Webberley, Associate Director, Analytical Research and Development at our sister company, SK biotek Ireland Limited, for his assistance with the profiles of AAS, ICP-OES, ICP-MS, and XRD.

Elemental impurities in food have been in the news recently, with reports of everything from lead being found in baby food to arsenic, cadmium, and assorted heavy metals in dark chocolate and other foods.1-8 Of course, the FDA and Congress are taking notice.3 The US Pharmacopeia definition of elemental impurities states they “include catalysts and environmental contaminants that may be present in drug substances, excipients, or drug products. These impurities may occur naturally, be added intentionally, or be introduced inadvertently (e.g., by interactions with processing equipment and the container closure system).”9 As recent news reports show, the concern can be expanded beyond drugs and APIs (active pharmaceutical ingredients) to include food and beverages…(Read More)
2/28/2023

Nitrosamines in Food and Beverages
This is the third in a series of entries examining nitrosamines in a range of products. Our first of two previous articles presented an overview of nitrosamines, including a historical look at their implication as a probable carcinogen. In the second entry, we reviewed their presence in active pharmaceutical ingredients (APIs), and how to remove them.
Nitrosamines are organic compounds found in the human diet and other environmental sources. These highly potent carcinogens can cause tumors in nearly all organs and have been classified as genotoxic impurities (GTI)… (Read More)
2/7/2023
 Nitrosamines in Active Pharmaceutical Ingredients
Nitrosamines in Active Pharmaceutical Ingredients
The linkage between cancer and a large class of chemical compounds known as nitrosamines were postulated by William Lijinsky in 1970.1 Then, in June 2018, their presence (specifically, N-nitroso-dimethylamine (NDMA)) was detected in the API Valsartan, an Angiotensin-II-receptor antagonist.
It later became “obvious that the issue may not only occur with sartans but, in principle, with any API containing a vulnerable amine and a nitrosation source. Hence not only NDMA but a plethora of potential nitrosamines could be created.”2 They have been subsequently detected in other medicines resulting in 250 product recalls, affecting more than 1400 lots.3,4 The cost of recalls could be high.5 APIs or their impurities can become nitrosated “during the later stages of the synthetic process of the drug product manufacturing or even while in the completed, packaged product.”6 … (Read More)
1/18/2023
Nitrosamines: An Overview

Nitrosamines are organic compounds found in the human diet and other environmental outlets. Being potent carcinogens that can cause tumors in nearly all organs, they have been classified as genotoxic impurities (GTIs). There are guidelines and rulings by various regulatory organizations, including the FDA, EPA, EMA, and the IARC (International Agency for Research on Cancer). Their presence and attendant concerns have been noted for many years. A.J. Gushgari and R.U. Halden wrote in Chemosphere, Nitrosamines were first proposed as “environmental carcinogens by William Lijinsky in 1970…